ARTICLE SUMMARY:
Last November, FDA issued a long-awaited, final guidance on its standards for overseeing human cell and tissue products. It is the latest move in an ongoing effort by the agency to wrap its arms around the complex, evolving, and controversial area of tissue-based biologics and, by extension, the newer, fast-growing field of regenerative medicine. An excerpt from Senior Writer Wendy Diller’s October 17 feature, “Regulatory Shifts for HCT/P Products Put Orthobiologics Manufacturers on the Spot.”
Under the new guidance, FDA collectively classifies human cell, tissue, and cellular-based products (HCT/Ps) into tiers based on risk benefit assessment: those that are designated as Section 361 products meet four carefully defined criteria and can proceed straight to market without requiring FDA review. Any products that fail to meet even one of these criteria are designated as Section 351 products and have to meet much higher regulatory standards (see Figure below).
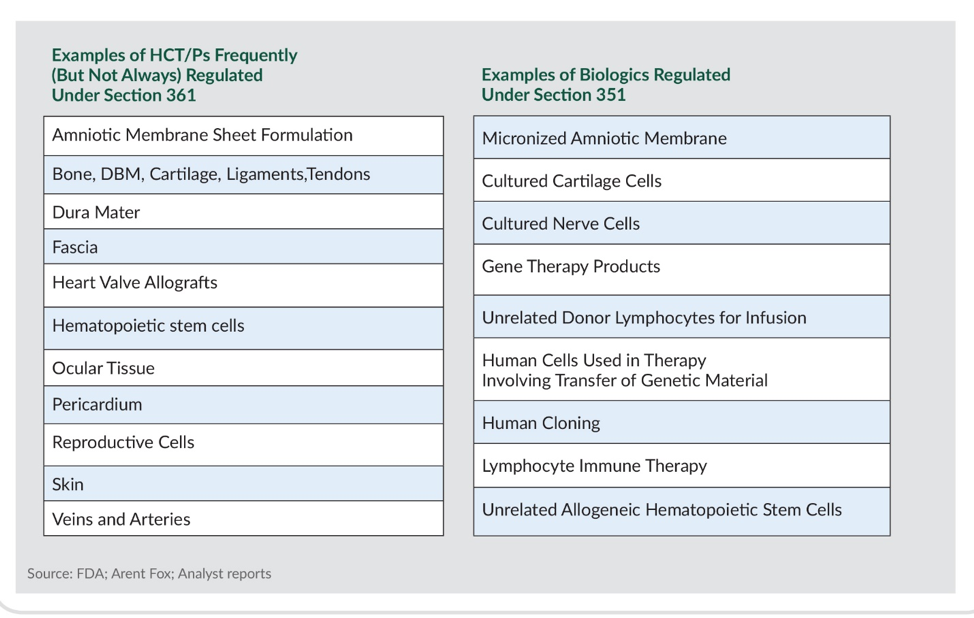
Depending on whether they defined as medical devices or biologics, the latter category must undergo a 510(k) or PMA process, or the even greater burden of a BLA review. Companies with products facing more rigorous review have a three-year window, which began in 2017, to fall in line.
As the FDA’s thinking evolved, it has left companies to self determine whether their products fall into the 361 or 351 camp, creating a lot of ambiguity and confusion. The 2017 regulations give industry much-needed direction on what kinds of products meet the criteria.
The distinctions have led many companies, which once saw tissue and cell therapies as an easy path to commercialization, now in the crosshairs of decision making.
The distinctions have led many companies, which once saw tissue and cell therapies as an easy path to commercialization, now in the crosshairs of decision making—i.e., whether to stick with only one of the two categories, invest heavily in bringing existing 361 products that are being shifted to 351 up to par while continuing to launch new products using higher standards of clinical evidence, or drop the whole field. Despite the latest guidance, which aims to further clarify the metrics used to assess each category’s eligibility, they are still somewhat confused and sorting out their strategies.
The changes have a particularly strong impact in orthopedics, where HCT/Ps are widely used for treating pain and facilitating post-surgical healing, especially when standard methods alone are insufficient. The large device manufacturers tend historically to view these products as a necessary part of the surgical armamentarium, but a minor influence on core growth, and thus they haven’t invested heavily in R&D that generates data on efficacy and differentiates them from competitors. The result is a commercial landscape full of me-too offerings, which lack strong clinical evidence of efficacy, and adhere to commodity dynamics. A handful of exceptions exist, but none for some of the biggest clinical gaps in orthopedic care, such as knee osteoarthritis (KOA).
The changes have a particularly strong impact in orthopedics, where HCT/Ps are widely used for treating pain and facilitating post-surgical healing, especially when standard methods alone are insufficient.
At stake are several subsectors of a multi-billion-dollar market, which covers cellular products derived from human tissue, such as amniotic membrane, adipose tissue, and bone, ligaments, skin, dura mater, heart valves, cornea, reproductive tissue, peripheral and cord blood, and more. The list includes demineralized bone matrix (DBM), stem cells derived from human tissue, including adipose-derived stem cells (but not those extracted from blood), and certain bone grafts. Excluded are products derived from blood, such as platelet-rich plasma (PRP), whole blood components, secreted human products, and minimally manipulated bone marrow.
All of this is on industry’s radar. “While companies have enjoyed the benefits of a low-regulation environment, they have understood the landscape is changing, and they need to prepare for a different regulatory world,” observes James Ravitz, the life sciences practice group leader at the law firm of Arent Fox, who has clients affected by the regulatory shifts.
Companies are conducting clinical trials and getting ready to file INDs for affected products, although it is not yet clear what steps FDA intends to take at the end of the three-year-window, Ravitz adds. FDA may not crack down immediately, but it reserves discretion to interfere if it is concerned about safety or efficacy, and it is risky for suppliers to wait until the last minute to start working on a product approval, he warns. For those now facing the prospect of filing BLAs, the evidence standards used are also ambiguous, as these many of these products have been on the market for years and are believed to be safe for clinical use. The agency applies a risk-benefit approach in judging how much safety and efficacy data are needed to satisfy regulatory approval requirements, he points out.
The regulatory shifts are a response in part to changing dynamics in the field, as it undergoes a renaissance due to advances in regenerative medicine, consumer hype, and an influx of investment. The opportunities for innovation are tempting, but the challenges are great.
Among the few suppliers willing to publicly discuss their intentions are MiMedx Group Inc. and Zimmer Biomet Inc. Both companies have responded by re-evaluating their biologics portfolios. MiMedx is under a cloud for financial reporting and auditing irregularities and faces potential delisting of its stock by NASDAQ—as a fallout, this summer it terminated for cause four of its top executives, including the founder, CEO, and chairman, Parker Petit, and brought in a new top management team. Senior executives insist, however, that these irregularities are not impacting its biologics development program.
#MedicalDevice #CommunityBlog #strategy #medtech #medicaldevices #ICYMI #trends #perspective #MedTechStrategist #global #orthobio #orthobiologics #regulatory #technology #fda #stemcell #manufacturers #mimedx #zimmerbiomet #regenerativemedicine #humancell #tissue #cellularbasedproducts #humantissue #FoodandDrugAdministration #NASDAQ
 Trial MyStrategist.com and unlock 7-days of exclusive subscriber-only access to the medical device industry's most trusted strategic publications: MedTech Strategist & Market Pathways. For more information on our demographics and current readership click here.
Trial MyStrategist.com and unlock 7-days of exclusive subscriber-only access to the medical device industry's most trusted strategic publications: MedTech Strategist & Market Pathways. For more information on our demographics and current readership click here.