
ARTICLE SUMMARY:
In this week’s roundup: Seema Verma's CMS is active in medtech, with key proposals on dialysis devices, telehealth, and mitral valve repair. Also, a global policy sweep, France’s first MDR notified body, and more.
Medicare is running a bit behind on its annual payment policy proposals this year, but they are starting to roll out, and, so far, dialysis technology and telehealth are two areas of impact. That and much more in this week’s Pathways’ Picks.
 Medicare Drafts
Medicare Drafts
Home dialysis bonus. Home dialysis systems could qualify for a recently created Medicare bonus payment program for dialysis devices under a July 6 CMS proposal. The provision is included in the 235-page 2021 End Stage Renal Disease Prospective Payment System proposed rule. Last year’s rule established the “transitional add-on payment adjustment for new and innovative equipment and supplies” (TPNIES), intended to facilitate access to new renal dialysis technology, but capital-related home dialysis machines were not included in its scope. The proposed TPNIES expansion fits into CMS’ broader efforts launched last year to expand adoption of home dialysis. (See “Advancing American Kidney Health: Will a New Executive Order be a Game-Changer for ESRD?” Market Pathways, August 19, 2019.)
“CMS data shows that those with ESRD have the highest rate of hospitalization of any single group, a trend consistent with having to frequently leave home to receive dialysis,” CMS Administrator Seema Verma said July 6. “Today’s action represents a sorely needed course direction [sic], making it easier for ESRD facilities to make new and innovative home dialysis machines available to patients who need them.”
The extra TPNIES payments are set at 65% of the Medicare Administrative Contractor (MAC)-determined price. To qualify, a device manufacturer needs to show its technology substantially improves diagnosis or treatment of Medicare beneficiaries relative to previously available options. CMS is also proposing a few other administrative reforms to the TPNIES program, including officially setting three years after FDA authorization as the cut-off point of when a device can no longer be considered sufficiently “new” to qualify.
 Tough reviews for Baxter, Outset. Meanwhile, the experience so far of the first two applicants to the program demonstrates that qualifying for a TPNIES payment will be no easy task. Feedback on add-on payment applications from Baxter International Inc. and Outset Medical from the CMS TPNIES Work Group summarized in the ESRD proposed rule suggests both companies are facing an uphill climb.
Tough reviews for Baxter, Outset. Meanwhile, the experience so far of the first two applicants to the program demonstrates that qualifying for a TPNIES payment will be no easy task. Feedback on add-on payment applications from Baxter International Inc. and Outset Medical from the CMS TPNIES Work Group summarized in the ESRD proposed rule suggests both companies are facing an uphill climb.
Baxter’s Theranovo 400 and 500 dialyzers, pending an FDA de novo classification, are a promising technology, according to the proposal. But, it notes, the Work Group “has concerns that the current state of evidence insufficiently demonstrates [substantial clinical improvement] in Medicare patients undergoing dialysis” (although, it suggests more evidence could be submitted during the comment period).
Outset is seeking an add-on payment for its Tablo Cartridge, which is a component of its recently cleared home hemodialysis system. In this case, CMS cites a policy complication—the cartridge itself is not a new device because it is already used in the more established, in-facility Tablo hemodialysis system. But Outset can’t seek a TPNIES for the redesigned home system because capital-related home dialysis systems don’t currently qualify. That’s an issue that would be addressed by this year’s proposed reforms, but the proposed rule suggests it could be a barrier for Outset for now.
Retaining telehealth allowances. The dialysis proposed rule came a week-and-a-half after CMS issued the first of CMS’ calendar-year payment rules—the 2021 Home Health proposed payment regulation, posted June 25. One medtech-noteworthy provision in that document is the proposal to permanently finalize the COVID-19 emergency provisions established in March allowing telehealth under the Medicare home health benefit, with certain restrictions. This is only one of a broader set of telehealth flexibilities that CMS has tagged to the ongoing pandemic, but it is one sign that many restrictions that have been loosened during the public health emergency could stay that way. Device trade group AdvaMed joined 340 health organizations in signing a June 30 letter calling on Congress to make all COVID-19 telehealth flexibilities permanent.
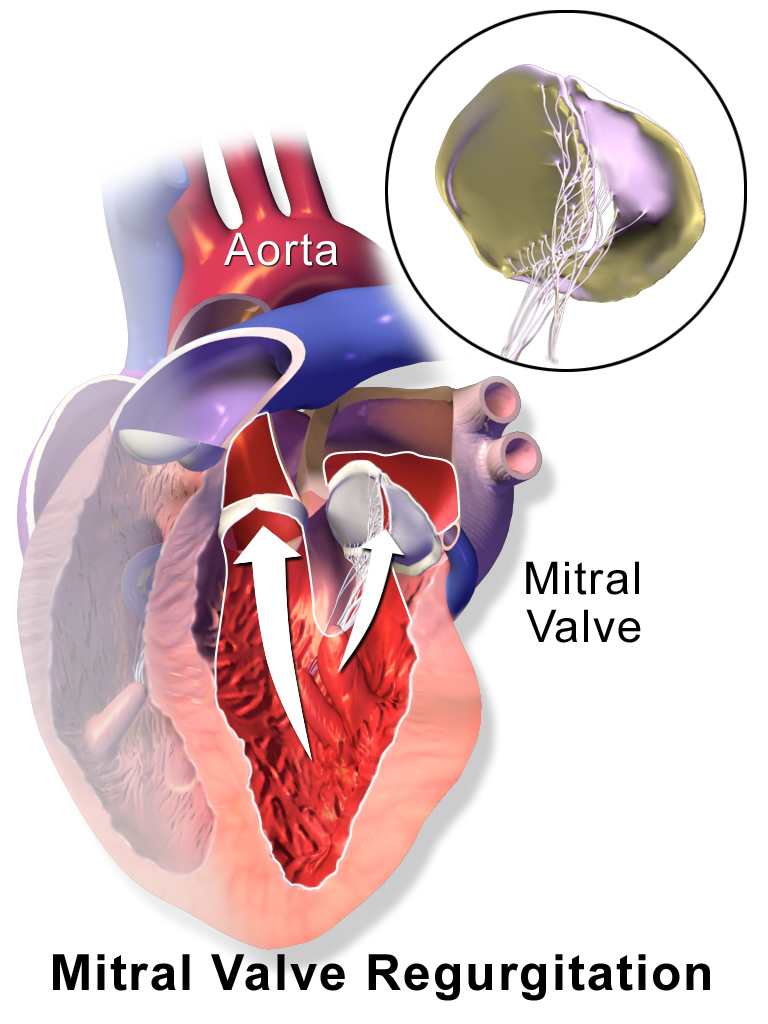 Mitral valve repair. Abbott Laboratories Inc. was delivered a win in a June 30 CMS proposal to expand coverage of edge-to-edge transcatheter mitral valve repair to treat functional mitral regurgitation in alignment with FDA’s March 2019 approval of Abbott’s MitraClip device for that indication. The proposal, which had been delayed for months, stipulates an array of conditions regarding the care team that must perform the procedure and the amount of experience necessary. The proposal would also remove existing coverage-with-evidence-development requirements for treating less-common degenerative MR, instead passing it off to regional contractors to make coverage decisions for that indication. Of note, CMS is proposing to change the name of the coverage policy from “Transcatheter Mitral Valve Repair (TMVR)” to “mitral valve Transcatheter Edge-to-Edge Repair (TEER)” to more precisely define the treatment addressed by the policy. That title addresses MitraClip, as well as some other in-development devices, such as Edwards Lifesciences Corp’s Pascal, but other types of technologies, including mitral valve replacement devices would likely require a new coverage policy. Edwards and Boston Scientific Corp., among others, had urged CMS to make the existing policy more flexible so new technologies could more seamlessly gain coverage upon FDA approval.
Mitral valve repair. Abbott Laboratories Inc. was delivered a win in a June 30 CMS proposal to expand coverage of edge-to-edge transcatheter mitral valve repair to treat functional mitral regurgitation in alignment with FDA’s March 2019 approval of Abbott’s MitraClip device for that indication. The proposal, which had been delayed for months, stipulates an array of conditions regarding the care team that must perform the procedure and the amount of experience necessary. The proposal would also remove existing coverage-with-evidence-development requirements for treating less-common degenerative MR, instead passing it off to regional contractors to make coverage decisions for that indication. Of note, CMS is proposing to change the name of the coverage policy from “Transcatheter Mitral Valve Repair (TMVR)” to “mitral valve Transcatheter Edge-to-Edge Repair (TEER)” to more precisely define the treatment addressed by the policy. That title addresses MitraClip, as well as some other in-development devices, such as Edwards Lifesciences Corp’s Pascal, but other types of technologies, including mitral valve replacement devices would likely require a new coverage policy. Edwards and Boston Scientific Corp., among others, had urged CMS to make the existing policy more flexible so new technologies could more seamlessly gain coverage upon FDA approval.
 Global Guidance
Global Guidance
June policy doc inventory. China’s National Medical Products Administration was the most prolific in rolling out new medtech policy documents among global agencies in June. NMPA issued 20 medical device/diagnostic documents, including 14 draft and final guidance documents, as well as six solicitations to support future guidance, according to Market Pathways’ “Document Depot.” US FDA was second with 14 guidance documents, templates, and other releases last month, followed by the UK Medicines and Healthcare products Regulatory Agency with five. Overall, guidance documents accounted for the majority of June entries in Document Depot—40 of a total of 73. Health technology assessments were a distant second with six entries.
Broadening companion Dx labels in China. NMPA issued a draft guideline July 7 outlining the regulatory process for updating labeling of a companion diagnostic approved for use with a specific drug to include use with multiple similar drugs that target the same genetic mutation or other biomarker. Comments are due August 3. Also on July 7, NMPA issued a solicitation seeking input from industry to help develop a guidance on technical review requirements for personalized implants made via additive manufacturing. (Links in Chinese.)
Brazil COVID-19 Q&A. ANVISA issued an updated Q&A document (link in Portuguese) July 7 tohelp explain Brazil's regulation relaxing rules for the manufacture, import, and purchase of priority medical devices for COVID-19.
 EU Check-In
EU Check-In
France’s first notified body. This week, GMED became the 15th notified body, and the first from France, to be designated under the Medical Device Regulation. The move comes more than a month after the MDR was originally scheduled to activate, but now there is some potential for GMED, which has had a large client base under the device directives, to engage in MDR assessments (albeit in the midst of COVID-19 restrictions) before the postponed regulation applies next May. GMED was designated with a relatively robust scope, including the ability to perform assessments of active implantable devices, but there are a handful of gaps and conditions on its MDR scope of practice.
Notified body survey. The European Commission released results from a survey of 37 notified bodies on the NBs' capacity and turnaround time for conformity assessments of COVID-19 devices and diagnostics. The survey questioned readiness under both the legacy directives and the Medical Device Regulation, although a sizeable chunk of the bodies are not designated under the MDR.
EU COVID test database. The European Commission recently launched a public database of marketed and in-development COVID-19 diagnostics, as well as scientific literature on test methods and devices. The inventory is based on the Commission’s own literature and web searches, and is also open to voluntary submissions from test developers.
More to Watch
 NEST’s Next Stage. The National Evaluation System for health Technology Coordinating Center (NESTcc) announced its long-promised launch of “NEST 1.0” June 30. Up until now, NESTcc has been focused on running pre-determined “Test Cases” primarily to provide proof-of-concept for generating “high-quality real-world evidence for medical devices across the total product lifecycle.” NEST 1.0 represents the initiative’s availability to partner on RWE projects that are initiated by device manufacturers, or by patient groups, regulators, and payors. Companies, for instance, could commission the NESTcc RWE network to support label-expansion studies, post-market safety studies, objective performance criteria, among other uses. Payors, on the other hand, might work with NESTcc to create quality and outcome measures. “As we move to a digitized world for device evaluation and surveillance, NEST can serve as a vehicle to drive evidence generation and enable new models of oversight to better meet the needs of patients,” said Jeffrey Shuren, FDA device center director.
NEST’s Next Stage. The National Evaluation System for health Technology Coordinating Center (NESTcc) announced its long-promised launch of “NEST 1.0” June 30. Up until now, NESTcc has been focused on running pre-determined “Test Cases” primarily to provide proof-of-concept for generating “high-quality real-world evidence for medical devices across the total product lifecycle.” NEST 1.0 represents the initiative’s availability to partner on RWE projects that are initiated by device manufacturers, or by patient groups, regulators, and payors. Companies, for instance, could commission the NESTcc RWE network to support label-expansion studies, post-market safety studies, objective performance criteria, among other uses. Payors, on the other hand, might work with NESTcc to create quality and outcome measures. “As we move to a digitized world for device evaluation and surveillance, NEST can serve as a vehicle to drive evidence generation and enable new models of oversight to better meet the needs of patients,” said Jeffrey Shuren, FDA device center director.
Multi-function devices. FDA sent a final guidance on “multiple function devices” for White House review July 8, the last step before it is rolled out publicly. The document, which was issued in draft form in 2018, was triggered, in part, by a requirement in the 21st Century Cures Act that prohibits FDA from regulating non-“device” software functions in digital health products, although FDA is extending its policy to all products with “medical device" and non-device functions, regardless of whether software is involved.
“Intended use” debate resurfaces in US regulatory agenda. The Department of Health and Human Services rolled out its spring “unified agenda” a few months late on June 30, including several new medtech-relevant items and many backlogged regulations. Among the new additions: FDA signaled it is working on a proposed rule to revisit a controversial 2017 plan to broaden the definition of “intended use” for device and drugs. FDA redefined intended use and the evidence that can be used to determine it in a 2017 final rule, but that the agency decided to indefinitely delay that policy in the face of push back from industry, which said FDA was pursuing an unsupported legal standard.
Also new on the latest HHS agenda: a proposed rule to update “good guidance practices.” Meanwhile, some important projects that have appeared on previous agendas and still are in the works include: FDA’s ongoing effort to align the Quality System Regulation with the ISO 13485 standard and CMS’ effort to establish special coverage pathways for innovative devices.
 Combo COVID tests. FDA updated its Emergency Use Authorization templates for molecular SARS-CoV-2 tests to include information to support submissions for combination tests to differentiate SARS-CoV-2 from other respiratory organisms, including the flu. The update came in conjunction with the July 2 EUA for a combo flu-COVID-19 assay developed by the Centers for Disease Control & Prevention. FDA has previously authorized combo COVID-19 tests by bioMerieux Inc. and Qiagen. FDA also updated its EUA templates July 6 with more information to support tests for pooled patient samples.
Combo COVID tests. FDA updated its Emergency Use Authorization templates for molecular SARS-CoV-2 tests to include information to support submissions for combination tests to differentiate SARS-CoV-2 from other respiratory organisms, including the flu. The update came in conjunction with the July 2 EUA for a combo flu-COVID-19 assay developed by the Centers for Disease Control & Prevention. FDA has previously authorized combo COVID-19 tests by bioMerieux Inc. and Qiagen. FDA also updated its EUA templates July 6 with more information to support tests for pooled patient samples.
FDA roundup. Other recent updates from the US regulatory agency that came in just before or soon after the July 4 US holiday include:
- Another delay to enforcement of unique device identification requirements for Class I and unclassified devices until September 24, 2022, citing COVID-19 and other factors;
- A new Medical Device Development Tool was qualified by FDA to support development of artificial pancreas systems. It called the Insulin Dosing Systems: Perceptions, Ideas, Reflections, and Expectations (INSPIRE) Questionnaires
- FDA again updated its COVID-19 clinical trial guidance with more details on seeking informed consent and video conferencing.
