
ARTICLE SUMMARY:
For medtechs, the fastest path to market does not always equate to the largest revenues or the highest market share. By Lori Thompson, Red Team Associates.
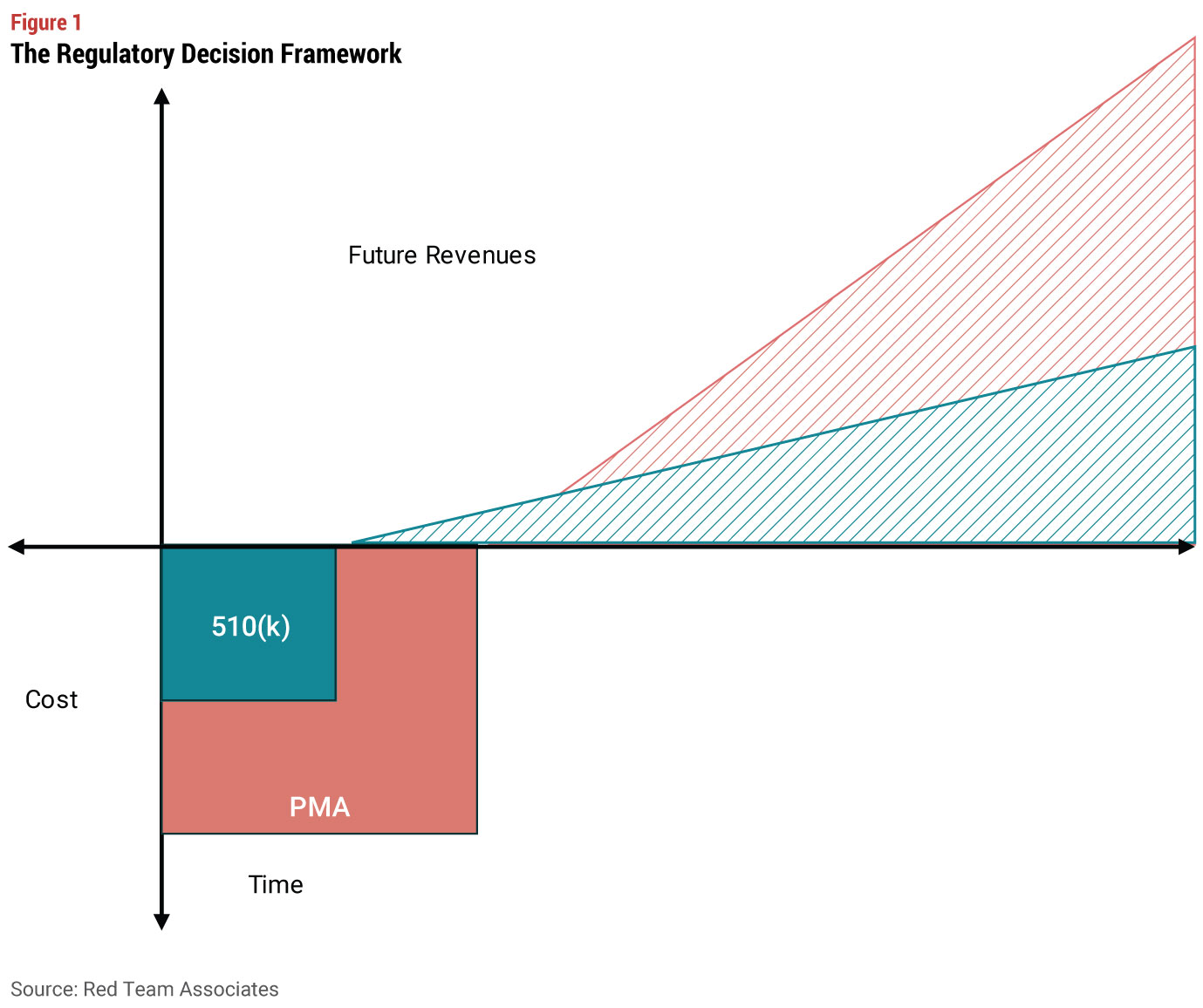
Typically, in the medical device field, we focus on the fastest path to market. We believe the adage “first to market wins.” While this has often been true, I would like to challenge our thinking. Sometimes the fastest path to market does not equate to the largest revenues or the highest market share.
|
510(k) Facts (1)
PMA W/out Panel Facts (2)
PMA with Panel Facts (2)
De Novo Facts (2)
|
I have been in many board rooms with medical device executives, reviewing product roadmaps, and heard someone say, “Forget it, we will never do a PMA.” Premarket Approvals are generally accepted as incredibly lengthy and expensive processes. As executives, we avoid this regulatory pathway at all costs, honoring the “speed to market” thinking.
A Changing Commercial Landscape Requires a Broader View of Regulatory Strategy
The commercial landscape has evolved. Medical product purchases are made based on data, especially in hospitals driven by Value Analysis Committees. (See “Are VACs Sucking the Life Out of Medtech Innovation? MedTech Strategist Community Blog, December 11, 2019.). These committees are looking for evidence of clinical benefit and significantly improved outcomes, modest improvements likely won’t overcome organizational inertia and hard switching costs. 510(k) devices innately have limited opportunities for differentiation, with claims and labelling based on an existing predicate device.
The regulatory process has also evolved. We commonly think about protecting our technologies by creating a portfolio of patents, investing in clinical studies, and establishing a sustainable cost structure with global manufacturing scale and sourcing. It’s time to think about the regulatory pathway as a critical part of our commercial strategy. A lengthier regulatory pathway could protect your product vs. future competition, and allow you to create a unique and compelling value proposition that maximizes your returns throughout the product life cycle.
I interviewed. David Feigal, MD, MPH, and a former Director at the Center for Devices and Radiologic Health (CDRH) within the FDA. Dr. Feigal has also served as the senior regulatory executive at Elan Pharmaceuticals and Amgen. He now works with NDA Partners, a regulatory consulting firm.
David has also experienced this active avoidance of the PMA process in many device companies, “It’s as if a drug company said ‘we don’t want to develop any new drugs, we just want to develop generics.’ It doesn’t make any sense as a strategy.”
510(k) Review Times are Increasing
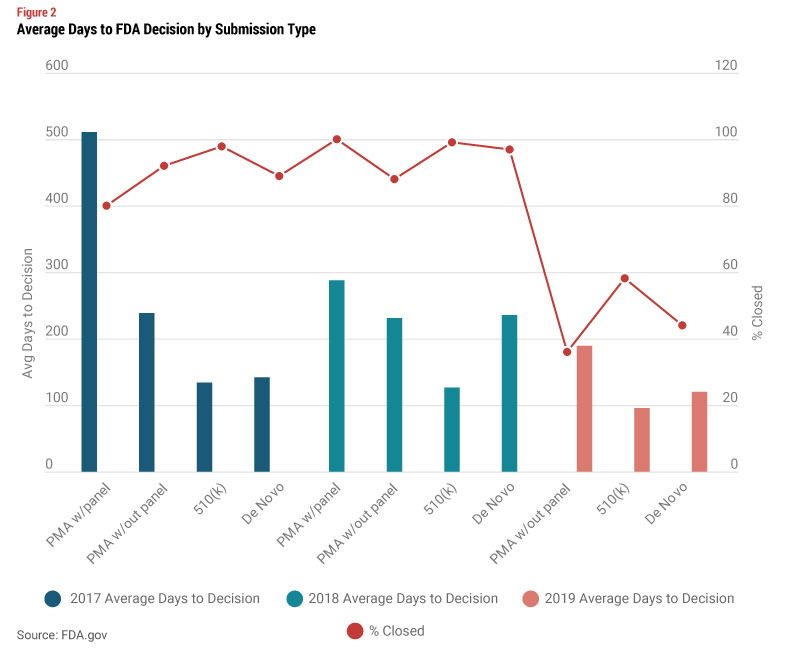
510(k) review times and requirements have in many cases increased, at least in part due to the spike in devices recalled in the last few years that entered the market through the 510(k) process.
Looking at FDA’s data on review times by submission type for the last three years, we see that on average, a PMA that did not require a panel review was about 100 days longer then a 510(k) review. If you are contemplating a regulatory strategy shift from a 510(k) to a PMA, it is not likely that your device fits into the “truly novel with no similar PMA category.” So a PMA without a review panel is the most likely alternative to a 510(k).
Planning Your Go-To-Market Roadmap
In your product go-to-market roadmap planning, ask yourself:
- Is it worth an extra hundred days to have significant competitive differentiation in your value proposition?
- Is it worth an extra hundred days to have novel indications for use or novel claims to support your messaging?
- Is it worth an extra hundred days to create a barrier to entry with a device not easily used as a predicate by a competitor?
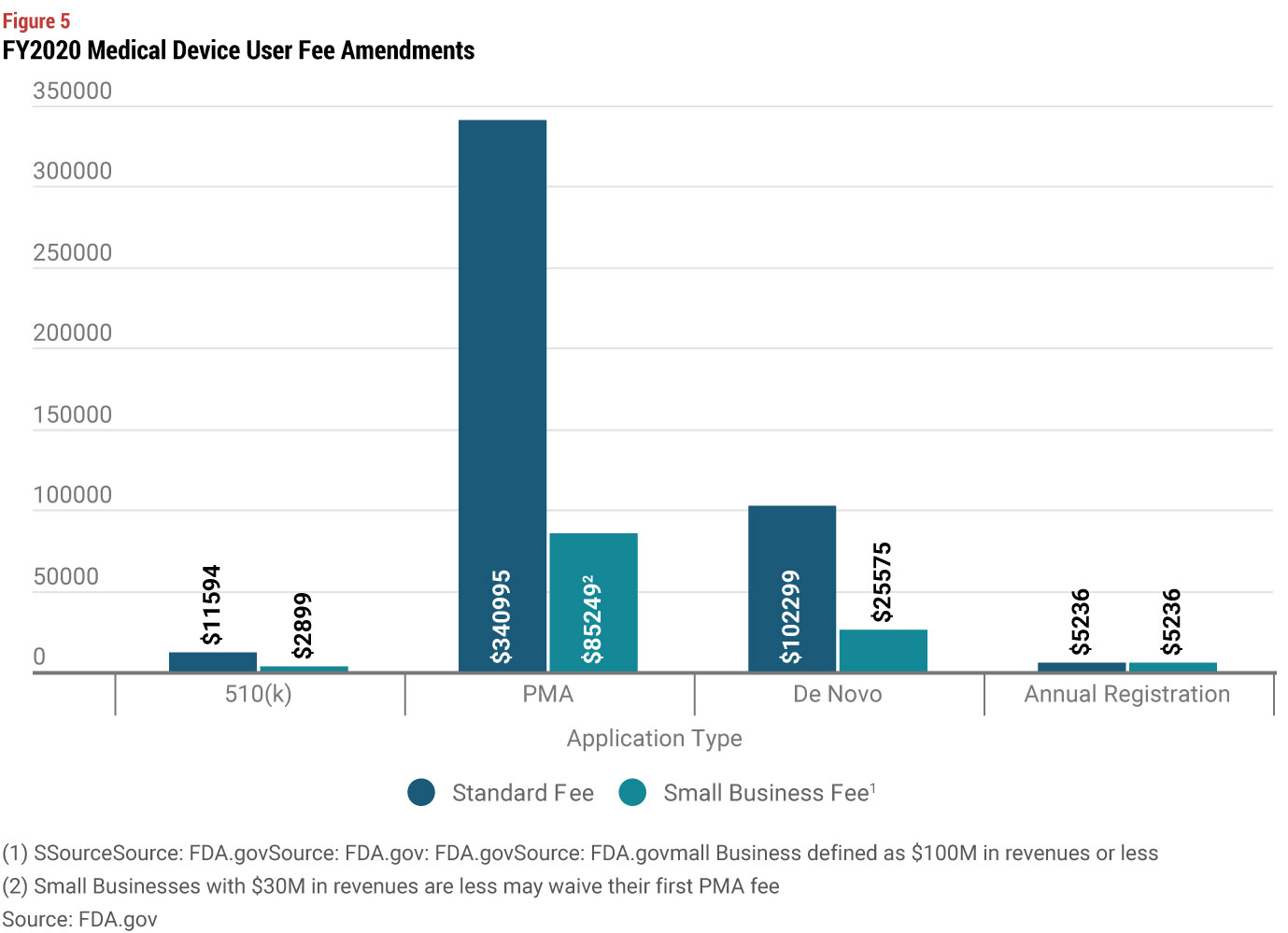
In answering these questions, you should also look at review times by type of device. Average review times for 510(k) have a smaller range of approximately 100 to 150 days depending on the type of device. But PMAs (without panel review) have a much bigger range, from approximately 100 to over 300 days. While you may conclude that taking a short term hit on launch timing is worth the benefit of creating more barriers to entry, you also need to consider the costs of a 510(k) vs. a PMA. FDA’s standard user fees are significantly higher for a PMA, but the difference in user fees is much more modest if you qualify as a small business (under $100 million in revenues). And for even smaller companies under $30 million in revenue, the user fee for your first PMA is waived.
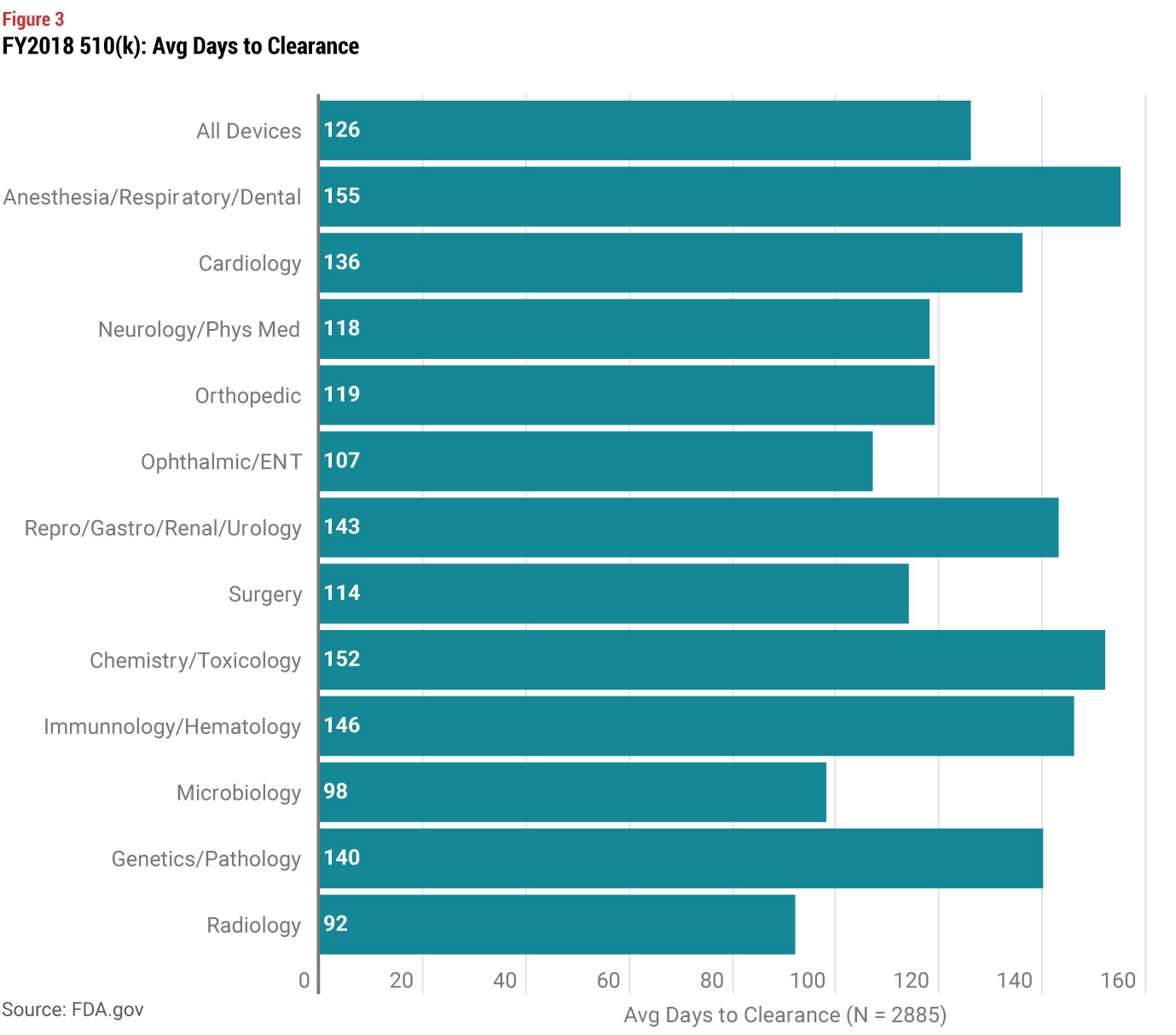
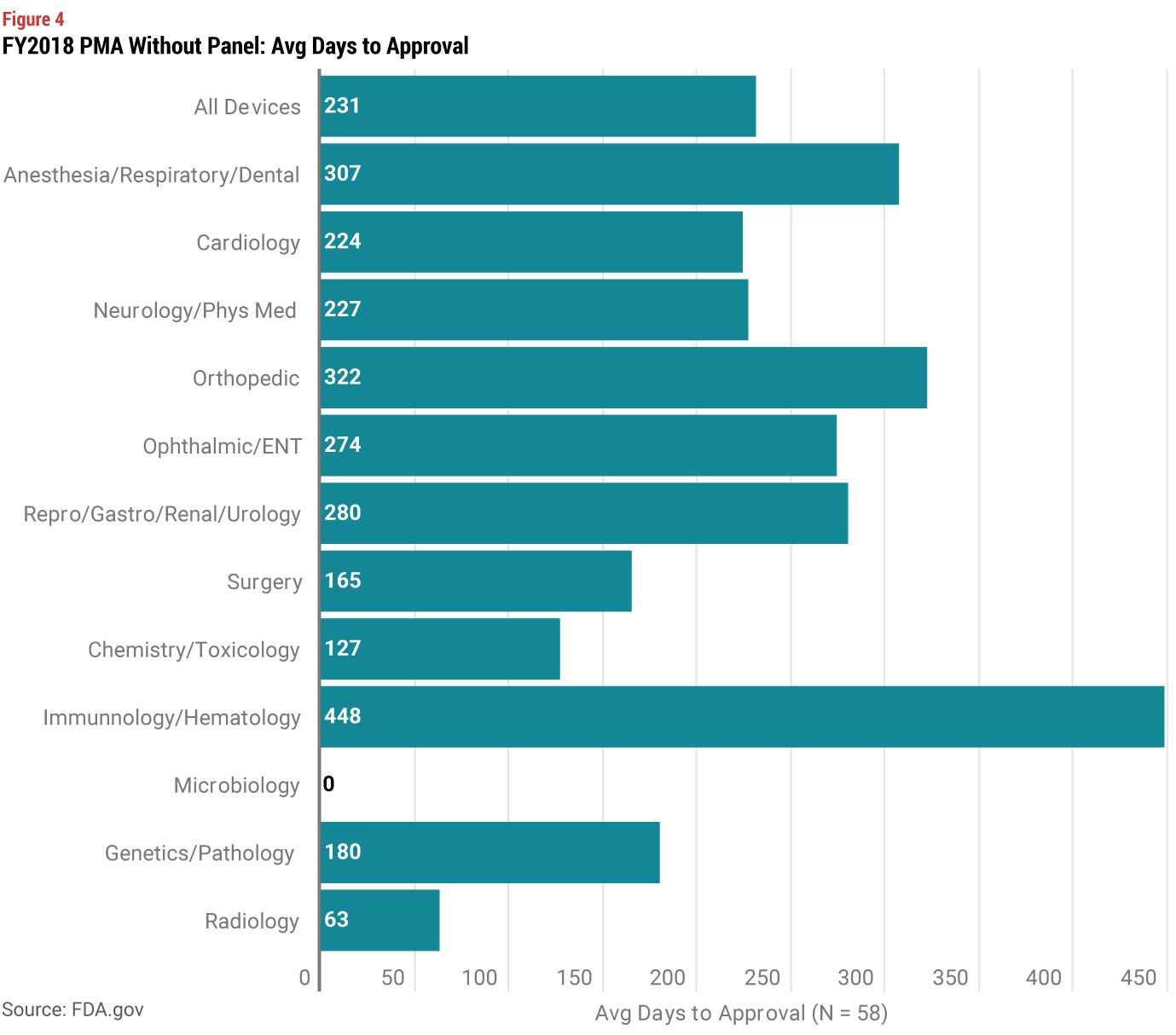
Is a PMA Worth the Time and Cost?
Of greater consideration is the difference in internal and external costs to prepare the required elements for a 510(k) vs. a PMA. Most (but not all) PMAs require clinical data. Most (but not all) 510(k)s do not require clinical data. This part of the process can be time consuming and very costly. But in thinking through the cost-benefit trade-offs, consider the following: what supporting data do you need to gain market acceptance and uptake? Medical device purchases are no longer driven primarily by physician preference. Significant device decisions are made by committee, taking into account the relative cost, patient outcomes, and relative risk of the new device. In other words, you need clinical evidence to establish market uptake.
Can you craft a clinical trial for your PMA submission that gives you compelling data supporting your value proposition? If yes, there may be a great benefit to launching with that data vs. back tracking and developing it later. Maybe the new adage should be “first to market with the strongest value proposition wins.”
Dr. Feigal brought to my attention an additional possible benefit for a PMA vs. a 510(k) device. He has had clients who “chose to take the PMA pathway primarily because it offered better chance of a higher reimbursement.” There is little data available on how CMS makes decisions on reimbursement levels. However anecdotal evidence suggests higher reimbursements are achieved for PMA devices vs. other regulatory pathways. Higher reimbursement levels can be critical to speeding up market uptake and creating overall higher returns on your investment.
De Novo and Multi-Tiered Strategies are Another Option
An historically underused submission pathway gaining traction is the de novo pathway. FDA created this option for Class I and Class II devices without an appropriate predicate device. FDA has made improvements to the review process for de novos, by no longer requiring a 510(k) submission prior to submitting a de novo request. Growth in de novo requests is also being fueled by a trend toward tightening definitions of acceptable predicate devices. De novos currently take about the same review time as a PMA without panel review, but the user fees and clinical study costs tend to be lower than a PMA. The downside of a de novo vs. a PMA is that de novos become immediately available as a predicate device for your competitors, unless you can craft indications or claims that a competitor cannot readily meet.
Another option to consider is a multi-layered strategy, where it is not 510(k) vs. de novo vs. PMA, but how you can create a product portfolio strategy utilizing multiple pathways. If establishing a beach head in a new market is critical, you can start with a 510(k) and get to market quickly. Then follow up with a unique set of indications and claims though a PMA or de novo submission. As Dr. Feigal put it, “Think about the full life cycle of your technology. You can start with a simpler version and gain some traction and experience, then move to a more complex version with more differentiated claims.”
|
Regulatory Strategy in Context of Your Strategic Goals Your regulatory strategy will be impacted not only by your commercial goals, but also by your longer-term strategic goals.
|
Commercial Strategy Should Influence Regulatory Strategy
The bottom-line is, when planning your commercial strategy for a new product, you should carefully review your regulatory options. A PMA pathway could allow you to:
- Create a unique value proposition supported by compelling clinical and outcomes data.
- Create additional protection for your device vs. competition.
- Allow your customers to access higher reimbursement levels.
- Create substantial value for your company and your investors.
Lori Thompson is Managing Director at Red Team Associates. She has more than 30 years of experience developing and commercializing new medtech products. Lori has held senior leadership positions in strategy and marketing with such leading companies as Tyco Healthcare, Cardinal Health, and CareFusion.
 Trial MyStrategist.com and unlock 7-days of exclusive subscriber-only access to the medical device industry's most trusted strategic publications: MedTech Strategist & Market Pathways. For more information on our demographics and current readership click here.
Trial MyStrategist.com and unlock 7-days of exclusive subscriber-only access to the medical device industry's most trusted strategic publications: MedTech Strategist & Market Pathways. For more information on our demographics and current readership click here.