
ARTICLE SUMMARY:
MedTech Europe's recent guideline tries to explain how companies can comply with a distinctive element of the EU’s impending unique device identification requirements. New software guidance from China and the UK, and CMS’ proposed new gapfill prices round out our June “Docs of the Month,” curated from Pathways’ Document Depot.
In Market Pathways’ “Docs of the Month” series, we spotlight key items from Pathways’ Document Depot—our running global inventory of medtech regulatory and policy documents—and provide useful details and context. This month, we dig into “MedTech Europe Guidance for Assigning Basic UDI-DI,” which was issued June 2 by the trade association MedTech Europe.
The concept of unique device identification (UDI) has become an increasingly familiar one to the device industry. Engaging with issuing agencies and being assigned UDIs that follow individual devices around, link the devices to online databases, and, policymakers hope, assist in tracking use, performance, and safety.
Since FDA finalized its UDI rule in 2013, the requirements have phased in over time and more and more companies have implemented UDIs. But now other countries have also taken steps toward implementing UDI programs, and while there is an overall goal of harmonization, new wrinkles are to be expected. Europe is now moving closer to implementing UDIs, as part of the Medical Device Regulation, and, in the case of the EU, that new wrinkle is called the Basic UDI-DI. And “Basic” does not necessarily mean simple.
MedTech Europe issued its guidance early in June attempting to bring together details from an array of sources to explain this feature, which will be unique to the EU system and central to the operation of its pending new and improved Eudamed device database. While other elements of the EU’s impending UDI system align with FDA and global approaches, the Basic UDI-DI is not required outside of Europe. Standard UDIs (or UDI-DIs, to represent the device, rather than production, elements of the unique code) are intended to be as precise as possible in identifying a device and particular attributes. It can follow a particular product through the supply chain. Basic UDI-DIs, on the other hand, are intended to have a broader sweep and more of an administrative function.
Specifically, a company is to use the Basic UDI-DI to connect all its devices with the same “intended purpose, risk class and essential design and manufacturing characteristics.” In general, a Basic UDI-DI can be associated with multiple UDI-Dis, but a UDI-DI can be associated with “one and only one” UDI-DI.
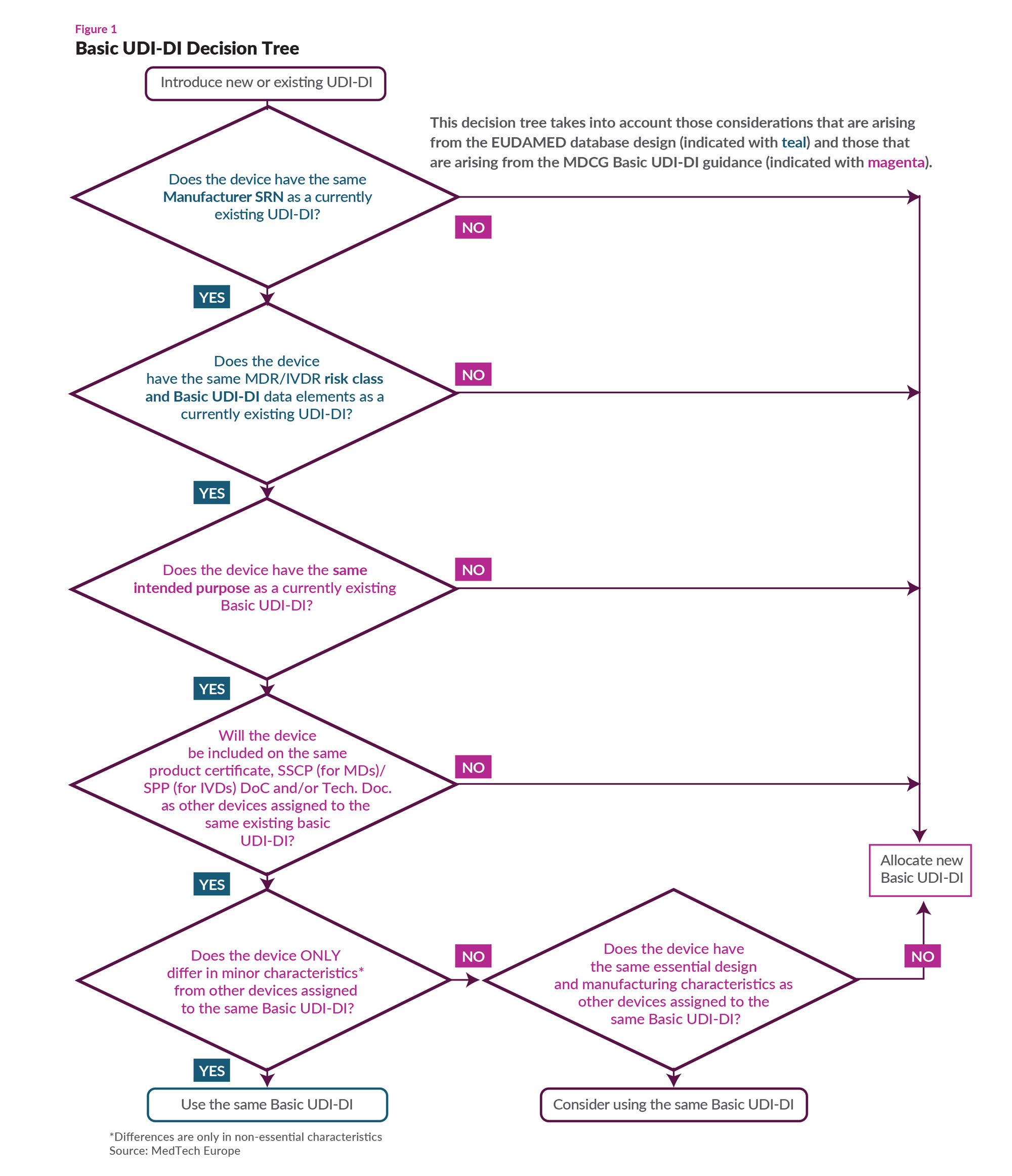 MedTech Europe’s new guidance principally concerns itself with what data elements are associated with a Basic UDI-DI and what considerations are necessary for deciding whether a device requires a new Basic UDI-DI or whether it fits within an existing one”(see Figure 1).
MedTech Europe’s new guidance principally concerns itself with what data elements are associated with a Basic UDI-DI and what considerations are necessary for deciding whether a device requires a new Basic UDI-DI or whether it fits within an existing one”(see Figure 1).
It will still be some time until Basic UDI-DIs can be fully employed as intended. The date of application of the MDR was recently postponed until next May, and the European Commission has said that Eudamed will not be fully operational until May 2022, aligning with when the IVDR is scheduled to apply. Recently, however, the Commission has said it will release certain database modules earlier, including the UDI/device registration module by the May 26, 2021, MDR application date. Whether the Commission meets that deadline or not, companies are still expected to seek Basic UDI-DIs and UDI-DIs for all products by the date of application for the regulations. MedTech Europe hopes its new guide will help.
Some other key documents captured in Pathways’ Documents Depot in June include:
Software guides: China’s National Medical Products Administration and the UK Medicines and Healthcare products Regulatory Agency both issued guidance documents on the regulation of software:
- “Regulatory status of software (including apps) used in the diagnosis, treatment and management of patients with coronavirus (COVID-19)” UK MHRA, June 5, 2020.
- “Guiding Principles for Technical Review of Medical Device Software (Second Edition Consultation Draft)” (Chinese), China NMPA, June 5, 2020.
Good reliance practices: The World Health Organization published a draft guidance June 8 outlining principles under which reliance on outside resources and consensus should be use as a tool for effective regulation and regulatory system strengthening: “Good reliance practices in regulatory decision-making: high-level principles and recommendations.”
Test payments: The Centers for Medicare and Medicaid Services issued Medicare Administrative Contractors’ proposed FY2020 "gapfill” prices for new lab test codes for public comment June 9: “2020 CLFS Gapfill Preliminary Determinations.”
 Trial MyStrategist.com and unlock 7-days of exclusive subscriber-only access to the medical device industry's most trusted strategic publications: MedTech Strategist & Market Pathways. For more information on our demographics and current readership click here.
Trial MyStrategist.com and unlock 7-days of exclusive subscriber-only access to the medical device industry's most trusted strategic publications: MedTech Strategist & Market Pathways. For more information on our demographics and current readership click here.